library(tidyverse)
library(patchwork)
# BiocManager::install(c("SingleCellExperiment","SingleR","celldex"),ask=F)
library(Seurat)
library(SingleCellExperiment)
library(SingleR)
library(celldex)Práctica 3: COVID-19 vs healhty
Biomarcadores y anotación de los tipos celulares
Descripción de los datos
Información general
- Título del estudio: Immunophenotyping of COVID-19 and Influenza Underscores the Association of Type I IFN Response in Severe COVID-19
- Especie: Homo sapiens
- Número de acceso de la base de datos GEO (Gene Expression Omnibus): GSE149689
- Tipo de experimento: scRNA-seq
Paso 1. Cargar paquetes y datasets en R
Cargar paquetes
Cargar el dataset de pbmc3k
# Cargar el objeto desde el archivo RDS
pbmc_integrated <- readRDS("output/pbmc3k_integrated.rds")
# Unir las capas del assay activo (ej. RNA)
pbmc_integrated <- JoinLayers(pbmc_integrated)
# Set the identity as louvain with resolution 0.5
sel.clust <- "RNA_snn_res.0.5"
# La identidad activa (active.ident) es la variable que Seurat usa por defecto para agrupar células en funciones como FindMarkers, DimPlot, etc.
alldata <- SetIdent(pbmc_integrated, value = sel.clust)
# Número de células en cada cluster
table(alldata@active.ident)
0 1 2 3 4 5 6 7 8
622 620 420 327 179 147 122 88 23 Número de células por dataset
table(alldata$orig.ident)
covid_1 covid_15 covid_16 covid_17 ctrl_13 ctrl_14 ctrl_19 ctrl_5
349 462 298 581 185 212 297 164 Paso 2. Identificación de genes marcadores
Corre FindAllMarkers para cada cluster en el objeto y devuelve una tabla con los genes marcadores de todos los clusters.
# Función: Identifica genes diferencialmente expresados entre dos grupos de células (ej. cluster 0 vs cluster 1, o cluster 0 vs todos los demás).
markers_genes <- FindAllMarkers(
alldata,
log2FC.threshold = 0.2, # umbral de cambio de expresión (default 0.25).
test.use = "wilcox", # método estadístico (ej. "wilcox", "MAST", "DESeq2").
min.pct = 0.1, # porcentaje mínimo de células que deben expresar el gen.
# Exige que la diferencia en la proporción de células que expresan un gen entre el grupo de interés y el grupo de referencia sea al menos del 20%.
min.diff.pct = 0.2,
only.pos = TRUE, # si TRUE, devuelve solo genes sobreexpresados.
max.cells.per.ident = 50, # Limita el número máximo de células usadas por cluster (ident) en el test de DE.
assay = "RNA"
)Calculating cluster 0Calculating cluster 1Calculating cluster 2Calculating cluster 3Calculating cluster 4Calculating cluster 5Calculating cluster 6Calculating cluster 7Calculating cluster 8markers_genes %>%
# Agrupa la tabla de genes marcadores por el cluster al que pertenecen.
group_by(cluster) %>%
# log2 fold change promedio sea mayor que 1.
dplyr::filter(avg_log2FC > 1) %>%
# Convertir en tabla
DT::datatable()
Note
Puedes ejecutar el mismo código anterior con otra prueba: «wilcox» (prueba de suma de rangos de Wilcoxon), «bimod» (prueba de razón de verosimilitud), «roc» (identifica «marcadores» de expresión génica mediante el análisis ROC), «t» (prueba t de Student), «negbinom» (modelo lineal generalizado binomial negativo), «poisson» (modelo lineal generalizado de Poisson), «LR» (regresión logística), «MAST» (modelo de vallas), «DESeq2» (distribución binomial negativa).
Important
Podrías pobrar cambiando la manera en la que se hizo el analisis tomando en cuenta lo siguiente y con la función FindMarkers:
- Clústeres grandes (0–3, >300 células): Wilcoxon.
- Clústeres medianos (4–7, 100–200 células): MAST o Wilcoxon.
- Clúster pequeño (8, 23 células): MAST o ROC, con interpretación cautelosa.
🧩 Interpretación: Para clústeres grandes (0–3) los resultados serán confiables con Wilcoxon; para clústeres medianos (4–7) puedes considerar MAST; y para el clúster 8, los resultados deben verse más como hipótesis que como evidencia sólida.
Paso 3. Visualizaciones de genes marcadores
Ahora podemos seleccionar los 25 genes más sobreexpresados para representarlos gráficamente.
top25 <- markers_genes %>%
group_by(cluster) %>%
top_n(-25, p_val_adj)
# Divide la ventana gráfica en una cuadrícula de 2 filas × 5 columnas, para mostrar hasta 10 gráficos en la misma figura.
par(mfrow = c(2, 5), mar = c(4, 6, 3, 1))
# for para los clusters
for (i in unique(top25$cluster)) {
barplot(sort(setNames(top25$avg_log2FC, top25$gene)[top25$cluster == i], F),
horiz = T, las = 1, main = paste0(i, " vs. rest"), border = "white", yaxs = "i"
)
abline(v = c(0, 0.25), lty = c(1, 2))
}
Heatmap
top5 <- markers_genes %>%
group_by(cluster) %>%
# Selecciona las 5 filas con el valor más pequeño de p_val_adj dentro de cada grupo.
slice_min(p_val_adj, n = 5, with_ties = FALSE)
# crear una ranura «scale.data» para los genes seleccionados
alldata_top5 <- ScaleData(alldata, features = as.character(unique(top5$gene)), assay = "RNA")Centering and scaling data matrixWarning: Different features in new layer data than already exists for
scale.data# save
# saveRDS(alldata_top5, file = "output/alldata_top5.rds")
# saveRDS(top5, file = "output/top5.rds")
# Visualizacion grafica
DoHeatmap(alldata_top5, features = as.character(unique(top5$gene)), group.by = sel.clust, assay = "RNA")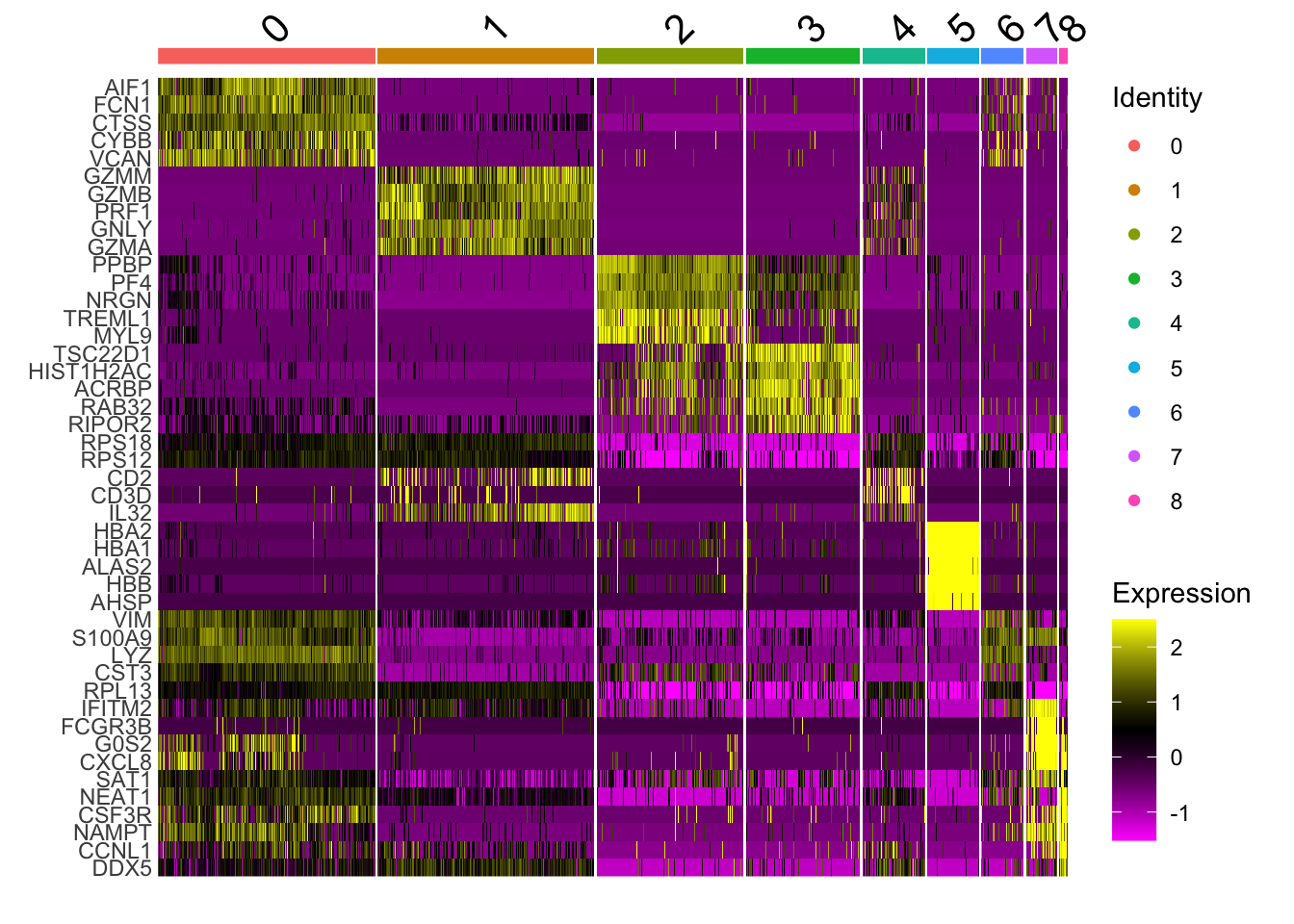
Dotplot
Otra forma consiste en representar las tasas globales de expresión y detección del grupo en un gráfico de puntos.
DotPlot(alldata_top5, features = rev(as.character(unique(top5$gene))), group.by = sel.clust, assay = "RNA") + coord_flip()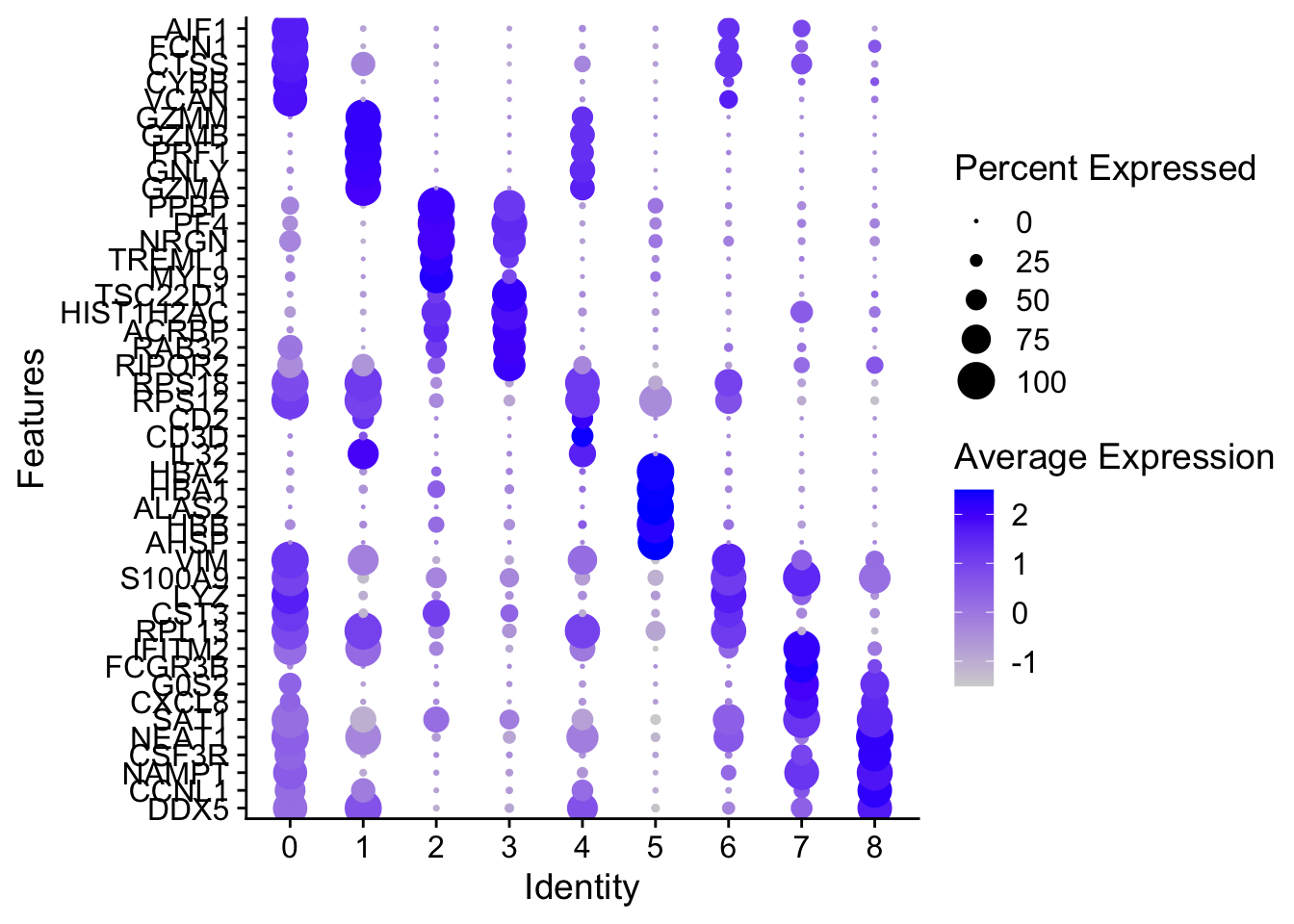
Violin plot
También podemos trazar un gráfico de violín para cada gen.
top3 <- markers_genes %>%
group_by(cluster) %>%
slice_min(order_by = p_val_adj, n = 3, with_ties = FALSE)
# saveRDS(top3, file = "output/top3.rds")
# Establece pt.size en cero si no quieres que todos los puntos oculten las formas del violín, o en un valor pequeño, como 0,1
VlnPlot(alldata_top5, features = as.character(unique(top3$gene)), ncol = 5, group.by = sel.clust, assay = "RNA", pt.size = 0)
Podemos visualizar genes característicos de las células Naive CD4+ T, como IL7R y CCR7, que se expresan de manera distintiva en este tipo de linfocitos. Estos genes funcionan como marcadores que nos ayudan a identificar y diferenciar esta población celular dentro del análisis de expresión.
VlnPlot(alldata_top5, features = c("IL7R", "CCR7"))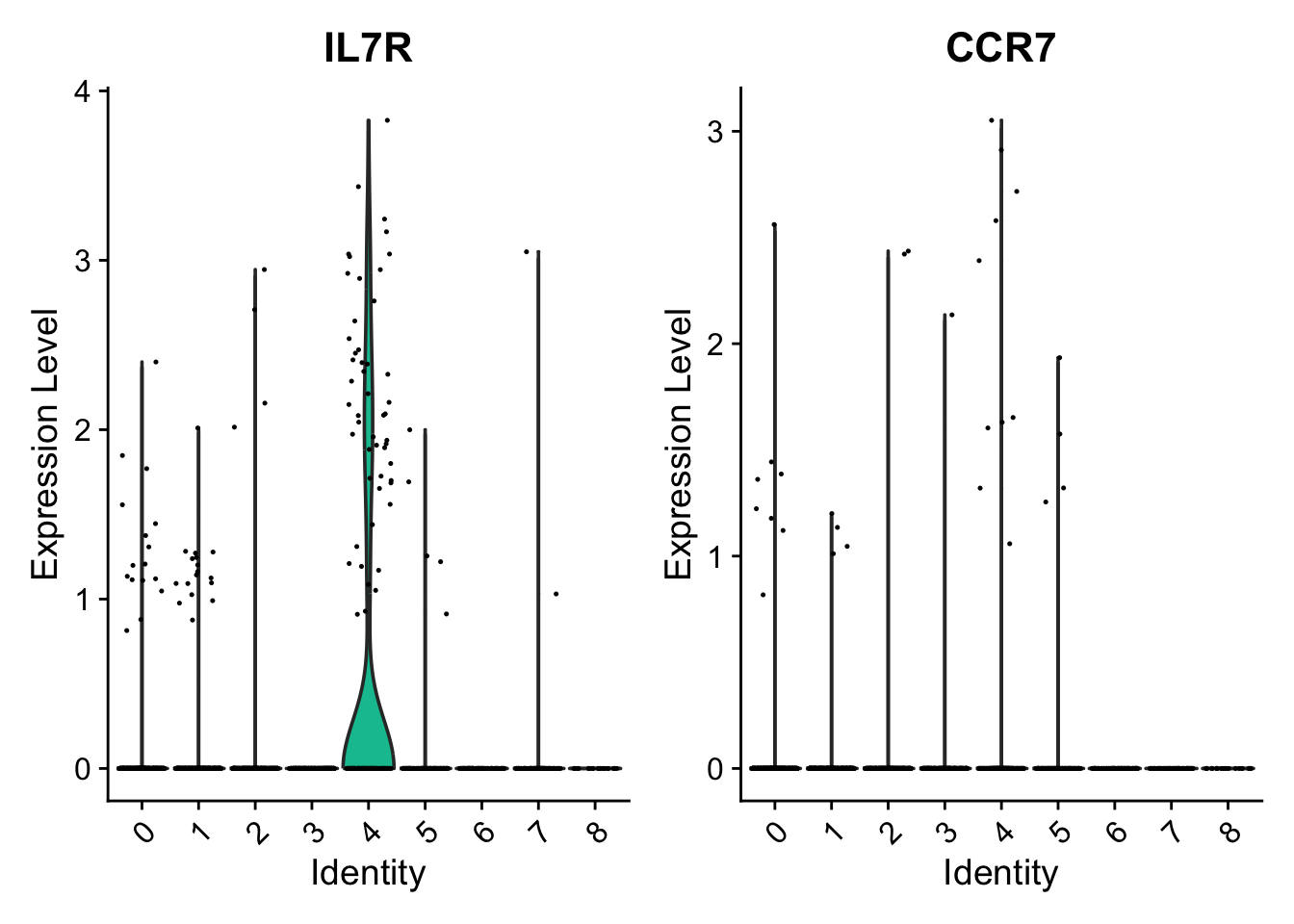
Gradientes en UMAP
Cada gen se proyecta como un mapa de calor sobre las coordenadas del UMAP, mostrando en qué regiones (clusters) se expresa más o menos.
📌 Qué vemos al observar estos genes
- MS4A1 (CD20) → marcador de linfocitos B. Se verá concentrado en el cluster de células B.
- GNLY (Granulysin) → marcador de células NK y citotóxicas. Se verá en clusters de NK/T CD8+.
- CD3E → marcador pan-T. Se distribuye en clusters de células T (CD4 y CD8).
- CD14 → marcador de monocitos clásicos. Se verá en el cluster monocítico.
- FCER1A → marcador de células dendríticas. Se verá en el cluster de DC.
- FCGR3A (CD16) → marcador de monocitos no clásicos y NK. Se verá en un cluster distinto de monocitos/NK.
- LYZ (Lisozima) → marcador de monocitos y granulocitos. Se verá en los clusters mieloides.
- PPBP → marcador de plaquetas. Se verá en el cluster plaquetario.
- CD8A → marcador de linfocitos T CD8+. Se verá en el cluster de T citotóxicas.
FeaturePlot(alldata_top5, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))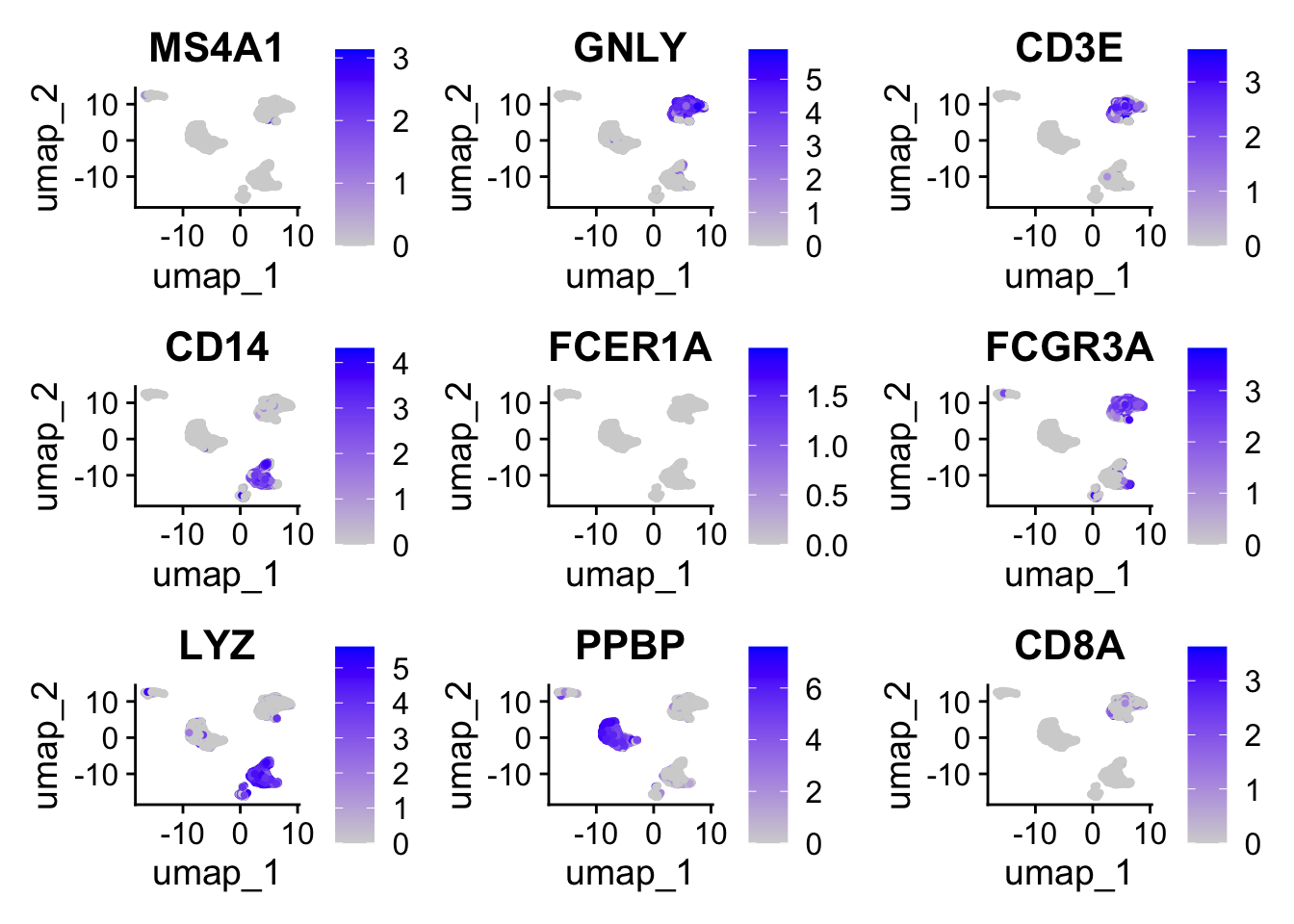
Paso 4. Asignar identidad celular a los clusters (opcional con Azimuth, problemas de paquetes)
En este dataset, cada cluster se puede asociar fácilmente a un tipo celular porque los genes diferenciales coinciden con marcadores bien establecidos.
En Azimuth, las columnas predicted.celltype.l1, predicted.celltype.l2 y predicted.celltype.l3 representan distintos niveles de detalle en la anotación automática de tipos celulares:
predicted.celltype.l1→ nivel más general (ej. “T cell”, “B cell”, “Monocyte”).predicted.celltype.l2→ nivel intermedio, con subtipos más específicos (ej. “Naive CD4 T”, “Memory CD4 T”, “CD14+ Monocyte”).predicted.celltype.l3→ nivel más fino, con anotaciones aún más detalladas según la referencia usada (ej. “Naive CD4 T (CCR7+)”, “Effector CD8 T”, “Plasmacytoid DC”).
library(Azimuth)
library(SeuratWrappers)
library(SeuratData)
library(GenomicRanges)
library(IRanges)
library(S4Vectors)
# Anotación automática usando referencia PBMC
pbmc_annotated <- RunAzimuth(
query = pbmc_integrated,
reference = "pbmcref"
)
# Ver anotaciones asignadas
head(pbmc_annotated@meta.data)Visualizaciones
# Cargar el objeto desde el archivo RDS
pbmc_annotated <- readRDS("output/pbmc3k_annotated.rds")
# Graficar UMAP con anotaciones automáticas
wrap_plots(
DimPlot(pbmc_annotated, group.by = "predicted.celltype.l1"),
DimPlot(pbmc_annotated, group.by = "predicted.celltype.l2"),
DimPlot(pbmc_annotated, group.by = "predicted.celltype.l3"),
ncol = 3
) + plot_layout(guides = "collect")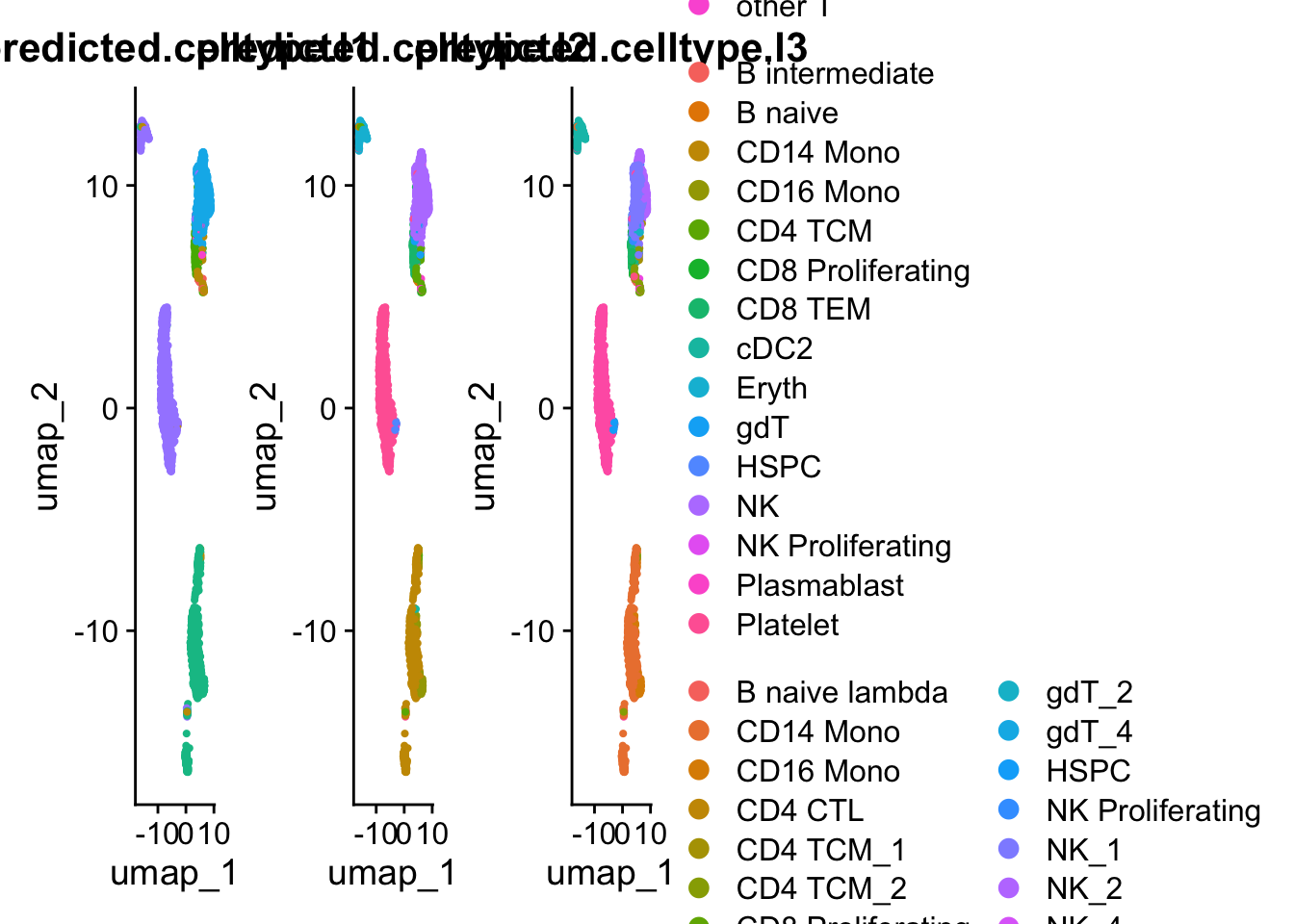
Paso 4. Asignar identidad celular a los clusters (SingleR + celldex)
# convertir de objeto seurat a SingleCellExperiment
sce <- as.SingleCellExperiment(pbmc_integrated)
sceclass: SingleCellExperiment
dim: 33538 2548
metadata(0):
assays(2): counts logcounts
rownames(33538): MIR1302-2HG FAM138A ... AC213203.1 FAM231C
rowData names(0):
colnames(2548): covid_1_AATAGAGAGGGTTAGC-1 covid_1_GGGTCACTCACCTACC-1
... ctrl_19_TTTACGTAGGTAGTCG-19 ctrl_19_TGTCCTGCAAATTGCC-19
colData names(12): orig.ident nCount_RNA ... seurat_clusters identreducedDimNames(3): PCA UMAP HARMONY
mainExpName: RNAaltExpNames(0):Ahora utilizaremos un paquete llamado SingleR para etiquetar cada célula. SingleR utiliza un conjunto de datos de referencia de tipos celulares con datos de expresión para deducir la mejor etiqueta para cada célula. El paquete celldex contiene una práctica recopilación de referencias de tipos celulares que, actualmente, incluye los siguientes conjuntos:
ls('package:celldex') [1] "BlueprintEncodeData" "DatabaseImmuneCellExpressionData"
[3] "defineTextQuery" "fetchLatestVersion"
[5] "fetchMetadata" "fetchReference"
[7] "HumanPrimaryCellAtlasData" "ImmGenData"
[9] "listReferences" "listVersions"
[11] "MonacoImmuneData" "MouseRNAseqData"
[13] "NovershternHematopoieticData" "saveReference"
[15] "searchReferences" "surveyReferences" En este ejemplo, utilizaremos el conjunto de datos «HumanPrimaryCellAtlasData», que contiene tipos de etiquetas tanto de alto nivel como de gran detalle. Descarguemos el conjunto de datos de referencia.
# descargar referencia de human cell atlas
ref.set <- celldex::HumanPrimaryCellAtlasData()Observar las labels que tenemos
unique(ref.set$label.main) [1] "DC" "Smooth_muscle_cells" "Epithelial_cells"
[4] "B_cell" "Neutrophils" "T_cells"
[7] "Monocyte" "Erythroblast" "BM & Prog."
[10] "Endothelial_cells" "Gametocytes" "Neurons"
[13] "Keratinocytes" "HSC_-G-CSF" "Macrophage"
[16] "NK_cell" "Embryonic_stem_cells" "Tissue_stem_cells"
[19] "Chondrocytes" "Osteoblasts" "BM"
[22] "Platelets" "Fibroblasts" "iPS_cells"
[25] "Hepatocytes" "MSC" "Neuroepithelial_cell"
[28] "Astrocyte" "HSC_CD34+" "CMP"
[31] "GMP" "MEP" "Myelocyte"
[34] "Pre-B_cell_CD34-" "Pro-B_cell_CD34+" "Pro-Myelocyte" Un ejemplo de los tipos de etiquetas «finas».
head(unique(ref.set$label.fine))[1] "DC:monocyte-derived:immature" "DC:monocyte-derived:Galectin-1"
[3] "DC:monocyte-derived:LPS" "DC:monocyte-derived"
[5] "Smooth_muscle_cells:bronchial:vit_D" "Smooth_muscle_cells:bronchial" Ahora etiquetaremos nuestras células utilizando el objeto `SingleCellExperiment`, con la referencia establecida anteriormente.
pred.cnts <- SingleR::SingleR(test = sce, ref = ref.set, labels = ref.set$label.main)Conserva los tipos que tengan más de 10 celdas en la etiqueta, vuelve a asignar esas etiquetas a nuestro objeto Seurat y visualízalas en nuestro umap.
lbls.keep <- table(pred.cnts$labels)>10
pbmc_integrated$SingleR.labels <- ifelse(lbls.keep[pred.cnts$labels], pred.cnts$labels, 'Other')
DimPlot(pbmc_integrated, reduction='umap', group.by='SingleR.labels')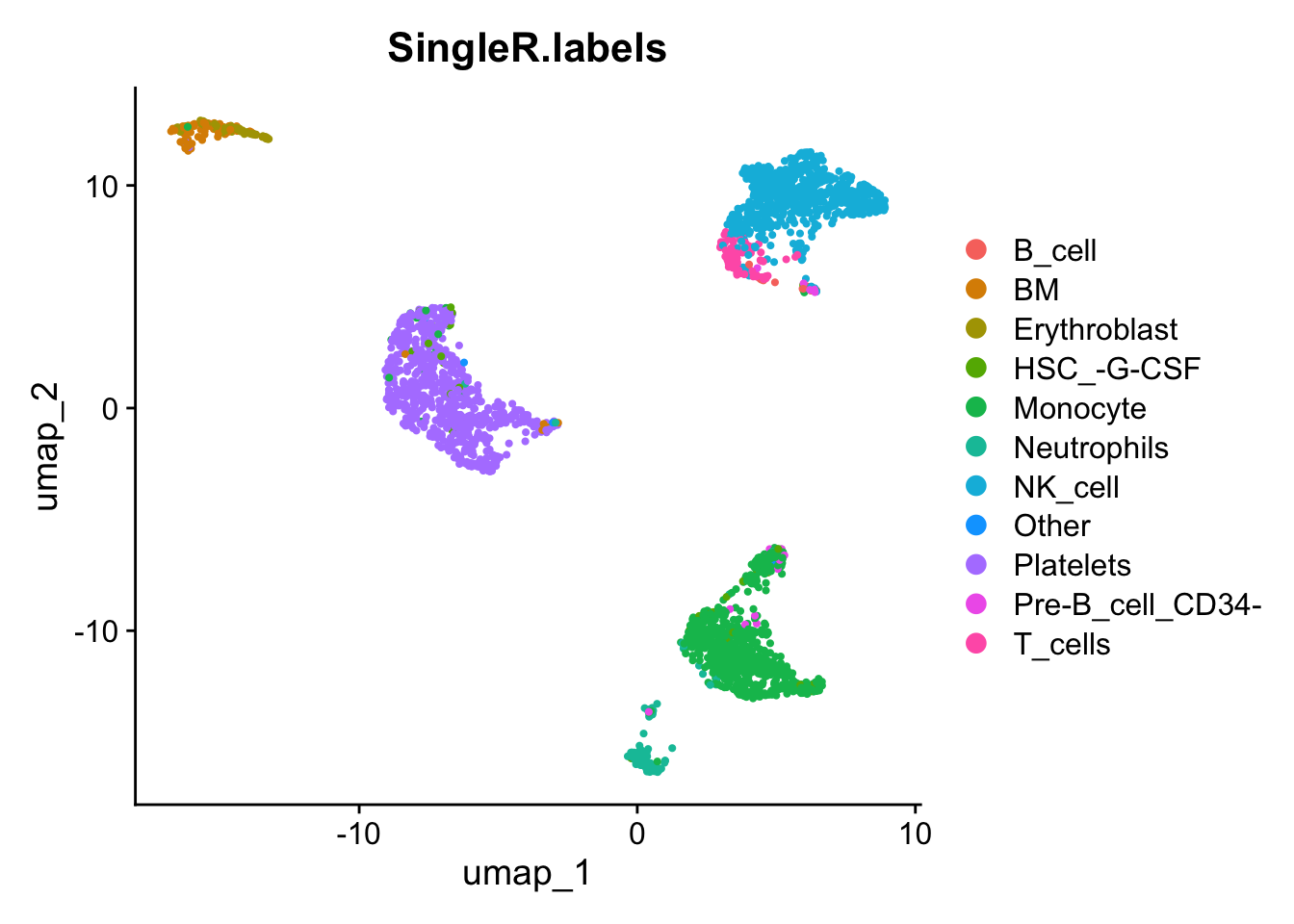
Es bueno ver que, aunque SingleR no utiliza los clústeres que calculamos anteriormente, las etiquetas parecen coincidir bastante bien con esos clústeres.
Paso 5. Marcadores conservados de tipos celulares
Para identificar marcadores conservados de tipos celulares en Seurat, se usa la función FindConservedMarkers(). Esta función busca genes que estén diferencialmente expresados en un cluster específico y que además sean consistentes entre condiciones o grupos (por ejemplo, entre diferentes donantes, tratamientos o lotes).
# Buscar marcadores conservados en el cluster 0
conserved_markers <- FindConservedMarkers(
pbmc_annotated,
ident.1 = 0, # cluster de interés
grouping.var = "type", # variable que define los grupos (ej. donantes)
assay = "RNA"
)Testing group Ctrl: (0) vs (1, 6, 4, 2, 5, 3, 8)Testing group Covid: (0) vs (2, 3, 5, 6, 1, 4, 7, 8)head(conserved_markers) Ctrl_p_val Ctrl_avg_log2FC Ctrl_pct.1 Ctrl_pct.2 Ctrl_p_val_adj
AC020656.1 8.594004e-143 4.743731 0.947 0.052 2.882257e-138
FCN1 5.480448e-124 3.441683 0.966 0.095 1.838033e-119
AIF1 2.837527e-123 3.454176 0.981 0.122 9.516497e-119
VCAN 5.693147e-110 3.704192 0.837 0.063 1.909368e-105
CYBB 3.026259e-142 4.819336 0.913 0.035 1.014947e-137
CSTA 3.932546e-126 3.976175 0.827 0.025 1.318897e-121
Covid_p_val Covid_avg_log2FC Covid_pct.1 Covid_pct.2
AC020656.1 2.292455e-270 3.989703 0.971 0.067
FCN1 1.089329e-265 3.913613 0.976 0.071
AIF1 3.147983e-260 3.824541 0.986 0.085
VCAN 1.391077e-257 3.809228 0.932 0.051
CYBB 5.556733e-256 3.455544 0.896 0.037
CSTA 2.601619e-251 3.517373 0.882 0.033
Covid_p_val_adj max_pval minimump_p_val
AC020656.1 7.688435e-266 8.594004e-143 4.584910e-270
FCN1 3.653392e-261 5.480448e-124 2.178659e-265
AIF1 1.055771e-255 2.837527e-123 6.295966e-260
VCAN 4.665394e-253 5.693147e-110 2.782154e-257
CYBB 1.863617e-251 3.026259e-142 1.111347e-255
CSTA 8.725310e-247 3.932546e-126 5.203238e-251Seleccionar los más relevantes
- Para encontrar marcadores conservados, lo más lógico es ordenar por
max_pval:- Si el máximo p‑valor sigue siendo bajo, significa que el gen es significativo en todos los grupos.
minimump_p_valsolo te dice el mejor grupo, pero no garantiza conservación.
top_conserved <- conserved_markers %>%
rownames_to_column("gene") %>%
arrange(max_pval) %>% # ordenar de menor a mayor
slice_head(n = 20) # tomar los 10 más robustos
head(top_conserved) gene Ctrl_p_val Ctrl_avg_log2FC Ctrl_pct.1 Ctrl_pct.2 Ctrl_p_val_adj
1 AC020656.1 8.594004e-143 4.743731 0.947 0.052 2.882257e-138
2 CYBB 3.026259e-142 4.819336 0.913 0.035 1.014947e-137
3 MNDA 2.470388e-129 4.278479 0.856 0.032 8.285188e-125
4 MPEG1 2.240135e-126 4.776329 0.798 0.018 7.512965e-122
5 CSTA 3.932546e-126 3.976175 0.827 0.025 1.318897e-121
6 FCN1 5.480448e-124 3.441683 0.966 0.095 1.838033e-119
Covid_p_val Covid_avg_log2FC Covid_pct.1 Covid_pct.2 Covid_p_val_adj
1 2.292455e-270 3.989703 0.971 0.067 7.688435e-266
2 5.556733e-256 3.455544 0.896 0.037 1.863617e-251
3 1.448351e-191 1.512578 0.973 0.114 4.857479e-187
4 5.200826e-246 4.456362 0.790 0.013 1.744253e-241
5 2.601619e-251 3.517373 0.882 0.033 8.725310e-247
6 1.089329e-265 3.913613 0.976 0.071 3.653392e-261
max_pval minimump_p_val
1 8.594004e-143 4.584910e-270
2 3.026259e-142 1.111347e-255
3 2.470388e-129 2.896702e-191
4 2.240135e-126 1.040165e-245
5 3.932546e-126 5.203238e-251
6 5.480448e-124 2.178659e-265Visualización
DotPlot(pbmc_annotated, features = top_conserved$gene, cols = c("blue", "red"), dot.scale = 8, split.by = "type") +
RotatedAxis()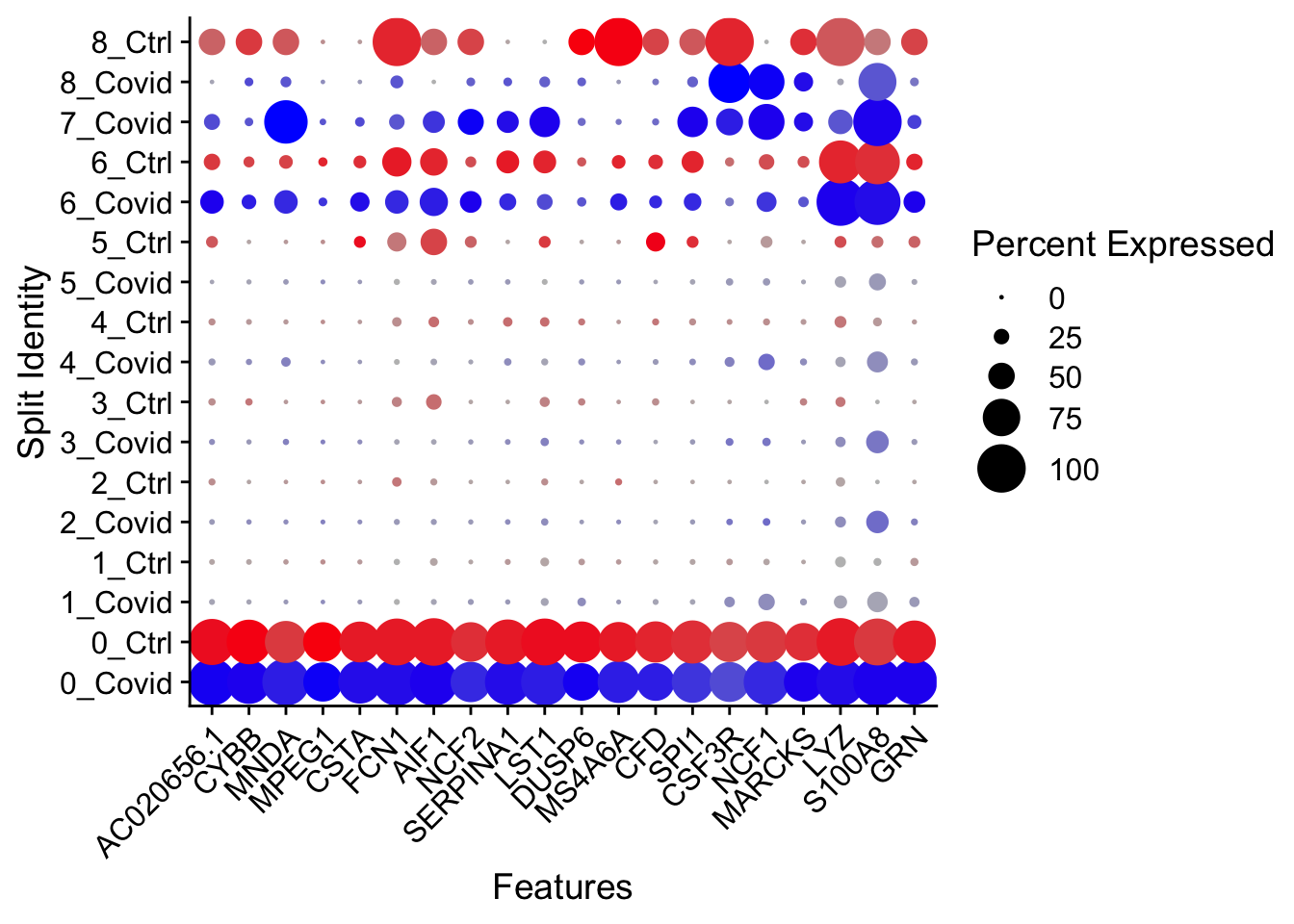
Interpretación 🔎
Si un gen aparece con puntos grandes y coloreados en ambos grupos (Control y Covid), significa que es un marcador conservado: se expresa de manera robusta en ese cluster sin importar la condición.
Si el color o el tamaño cambia mucho entre Control y Covid, ese gen podría ser un marcador diferencial entre condiciones, no conservado.
Paso 7. Guardar dataset
saveRDS(pbmc_annotated, file = "output/pbmc3k_annotated.rds")