7 Scripts en Bash
Los scripts son archivos de texto que contienen instrucciones ejecutables por un intérprete de un lenguaje de programación para la ejecución de tareas. El formato .sh se utiliza para escribir scripts ejecutables por un intérprete como Bash. En esta sección crearemos varios scripts de Bash y usaremos varios comandos y operadores.
Los script de Bash contiene al inicio esta descripción #!bin/bash y la extensión .sh
7.1 Actividad grupal
Paso 1. Crear un script en Bash con el nombre demo.sh empleando nano.
nano demo.shPaso 2. Copia el siguiente código dentro del script demo.sh y guarda los cambios.
#!/bin/bash
# File: demo.sh
# Input de archivo
# 2. Mostrar el archivo y número de argumentos
echo "Archivo a procesar > $@"
echo "Numero de argumentos: $#"
# 3. Conteo de secuencias y separación de malas lecturas
numero_secuencias=$(grep -c '^@SRR098026' $@)
grep -B1 -A2 NNNNNNNNNN $@ > malas_lecturas.fastq
malas=$(cat malas_lecturas.fastq | wc -l)
echo "Número de malas lecturas: $malas"
# 4. Búsqueda de patrones
echo "Desea buscar patrones (y/n): "
read d
if [[ $d == "y" ]]; then
echo "Los patrones se guardarán en: patrones.txt"
echo -e 'ACTG\nCCCCC\nNNNCNNN\nNNNGNNN\nTTTT\nTATA\nAAA' > patrones.txt
grep -f patrones.txt $@ > busqueda.txt
echo "Búsqueda de patrones guardada en: busqueda.txt"
else
echo "ok :P"
fi
# Mensaje final
echo "Fin :)"1. Comentarios y declaración de archivo
#!/bin/bash: Indica que el script se ejecutará usando el intérprete de Bash.# File: demo.sh: Un comentario que indica el nombre del archivo del script.2. Mostrar el archivo y número de argumentos
echo "Archivo a procesar > $@": Muestra el nombre del archivo que se pasará como argumento al script.$@representa todos los argumentos pasados al script.echo "Numero de argumentos: $#": Muestra cuántos argumentos ha recibido el script.$#indica el número de argumentos.3. Contar secuencias y separar malas lecturas
numero_secuencias=$(grep -c '^@SRR098026' $@): Usagreppara contar cuántas secuencias empiezan con@SRR098026en el archivo (probablemente sea un prefijo en un archivo FASTQ que identifica secuencias de interés).-ccuenta las coincidencias.grep -B1 -A2 NNNNNNNNNN $@ > malas_lecturas.fastq: Busca secuencias de calidad baja o problemáticas (que contengan 10 nucleótidos seguidos deN, que representan bases no determinadas).-B1: Muestra 1 línea antes de cada coincidencia deNNNNNNNNNN.-A2: Muestra 2 líneas después de la coincidencia.El resultado se guarda en un archivo llamado
malas_lecturas.fastq.
malas=$(cat malas_lecturas.fastq | wc -l): Cuenta el número total de líneas enmalas_lecturas.fastq(esto indica la cantidad de malas lecturas encontradas).echo "Número de malas lecturas: $malas": Muestra la cantidad de malas lecturas encontradasecho "Numero de argumentos: $#": Muestra cuántos argumentos ha recibido el script.$#indica el número de argumentos.4. Búsqueda de patrones
echo "Desea buscar patrones (y/n): ": Pregunta al usuario si desea buscar patrones específicos en el archivo.read d: Lee la respuesta del usuario y la guarda en la variabled.if [[ $d == "y" ]]; then: Si la respuesta es “y” (sí), el script procede a buscar los patrones.echo -e 'ACTG\nCCCCC\nNNNCNNN\nNNNGNNN\nTTTT\nTATA\nAAA' > patrones.txt: Crea un archivopatrones.txtque contiene una lista de secuencias de interés (patrones a buscar).grep -f patrones.txt $@ > busqueda.txt: Busca los patrones contenidos enpatrones.txtdentro del archivo de entrada. El resultado de la búsqueda se guarda enbusqueda.txt.echo "Búsqueda de patrones guardada en: busqueda.txt": Informa al usuario que los resultados de la búsqueda se guardaron en el archivobusqueda.txt.
else: Si la respuesta es “n” (no), simplemente muestra un mensaje.
Este código realizará las siguientes instrucciones:
Tomará como input el nombre del archivo
./_files/secuencias_bash.fastqde secuencias fastq para analizar.Buscará dentro del archivo provisto por el usuario las secuencias con el string @SRR098026 y las contará.
Exportará las malas secuencias a un archivo llamado
malas_lecturas.fastq, usando el string “NNNNNNNNNN” como plantilla.Reportará el número de malas secuencias.
Luego buscaremos patrones en el archivo
buenas_lecturas.fastq, si es que el usuario lo desea.
Paso 3. Deberías correr el script así, una vez que hayas descargado el archivo secuencias_bash.fastq:
bash demo.sh secuencias_bash.fastq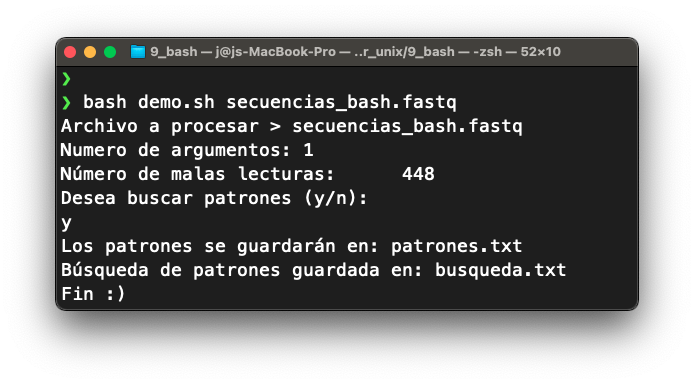
7.2 Repaso
Ahora que ya sabes realizar Script en Bash, podrias intentar realizar la actividad de 6. Extracción de información de archivos fastq del curso de GNU/Linuz para Bioinformatica organizado por RSG Ecuador.
7.3 Extracción de información de archivos fastq
En este ejercicio se obtendrá información de archivos fastq, que cotienen secuencias de nucleótidos y la calidad de su proceso de secuenciación. Para esto usaremos los archivos secuencias1.fastq, secuencias2.fastq y secuencias3.fastq que se encuentra en la carpeta _files dentro del directorio raíz del repositorio de GitHub unix.bioinfo.rsgecuador .
Ejercicio 6.1 La letra N representa un nucleótido que no pudo ser leído correctamente, y se reporta como ninguno, o missing data. Imprime el número de secuencias que tenga diez N seguidas por cada uno de los archivos fastq de la carpeta _files.
Es posible realizar esto con un comando de una sola línea, intenta resolverlo de esta forma.
Ejercicio 6.2 Ahora, determina el número de secuencias (lineas) de cada archivo .fastq por separado. Se conoce que los títulos de las corridas de las secuencias en cada archivo .fastq empiezan con el string @SRR098026.
7.4
Material suplementario
- RSG Ecuador. Scripts en Bash
- RSG Ecuador. Wildcards y Streams
- RSG Ecuador. Expresiones regulares (regex)
- Wildcard Selection in Bash