4 Análisis de datos de RNA-Seq
4.1 Bioinformática se conforma de la computación, biológica, matemáticas y estadística
La bioinformática, en relación con la genética y la genómica, es una subdisciplina científica que implica el uso de ciencias informáticas para recopilar, almacenar, analizar y diseminar datos biológicos, como secuencias de ADN y aminoácidos o anotaciones sobre esas secuencias NIH, 2023.
4.2 Transcriptoma
Es el conjunto de todas las moléculas de RNA producidos por el genoma bajo condiciones específicas o en una célula específica (scRNA-Seq) o en una población de células (bulk RNA-Seq).
4.2.1 ¿Porque es importante medir los cambios en la expresión génica (transcriptoma)?
Las variaciones en la expresión entre condiciones se puede relacionar con los cambios en los procesos biológicos.
El transcriptoma nos da una aproximación de los cambios relativos en la expresión génica de los genes codificantes y no codificantes.
- Genoma - Fijo
- Transcriptoma - Altamente variable
4.3 El transcriptoma varía según:
- Tejido / Órgano
- Célula
- Ambiente (estrés)
- Medicamentos (tratamientos)
- Salud
- Edad
- Etapa del desarrollo
4.4 Idea principal de RNA-Seq
4.4.1 Relacionar un fenotipo con los cambios de expresión de los genes en una condición dada
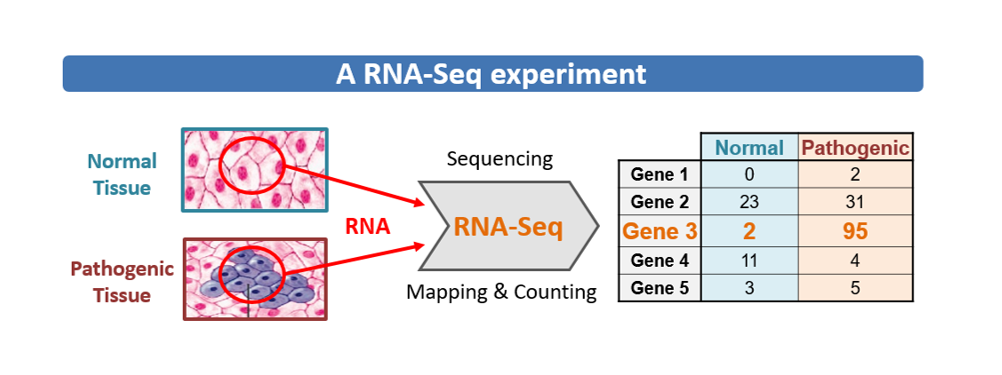
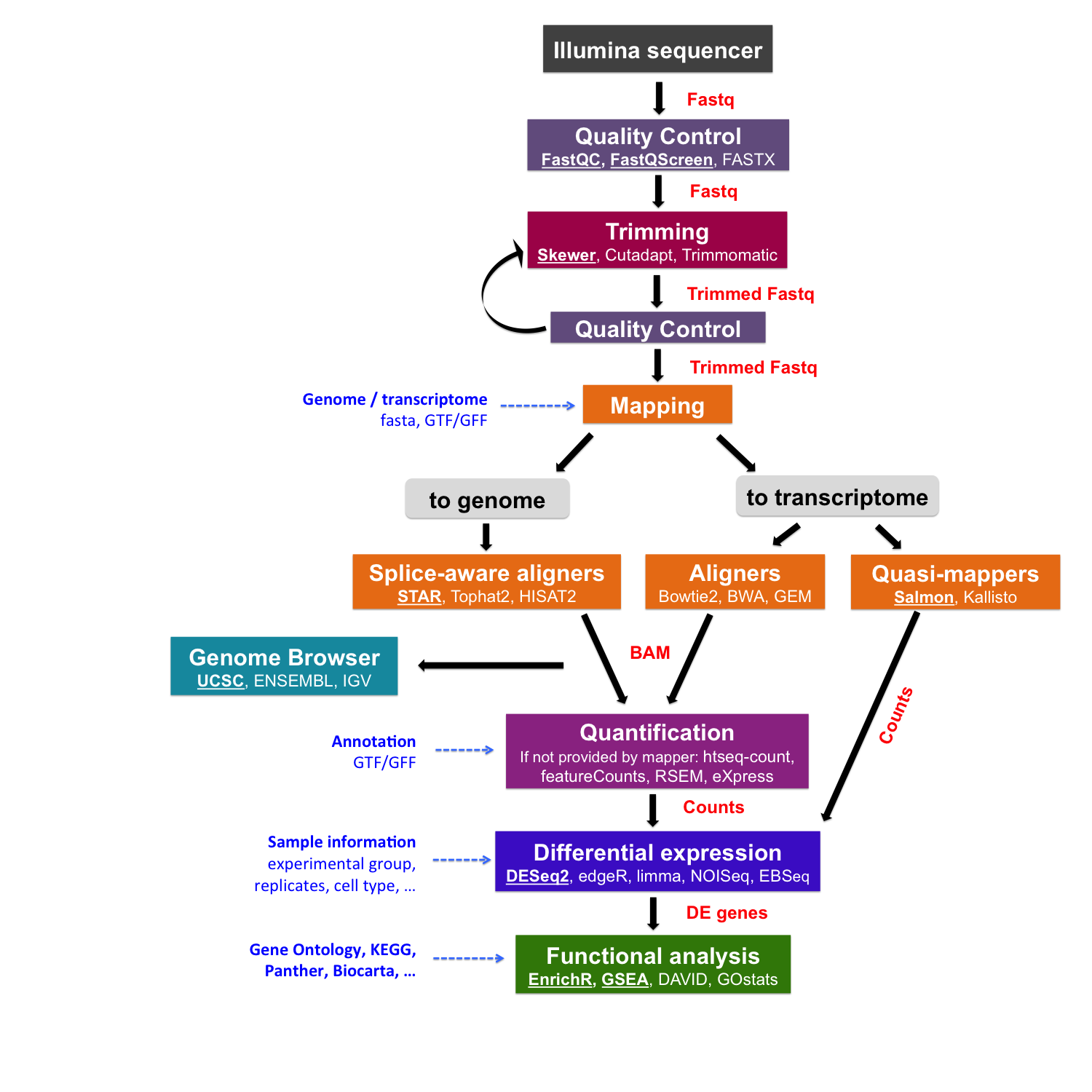
4.5 Pipeline general
5 Control de calidad de los datos
5.1 Quality Check / Quality Control
Uno de los pasos más importantes. Dedícale tiempo.
La calidad de tus datos importa, bibliotecas mal secuenciadas genera datos desconfiables.
Debes analizar su calidad para poder reclamar en la secuenciación (~1 semana).
5.2 Archivos fastq (fastq.gz / fq.gz)
Derivan del formato FASTA.
Muestra la calidad de cada nucleótido.
Cada secuencia está representada por 4 líneas:
@ ID del read + información de la corrida
Secuencia
Símbolo “+”
Información de la calidad de secuenciación de cada base. Cada letra o símbolo representa a una base de la secuencia codificado en formato (Escala Phred y código ASCII)

5.3 Phred Quality score / Puntuación de calidad
Q = -10 x log10(P) # where P is the probability that a base call is erroneous
Q = Phred Quality score
P = Probability of incorrect base call

El valor máximo de calidad es = ~40 (zona verde) y los valores < 20 se consideran de baja calidad.
Reporte de FastQC con buena calidad de las lecturas en la zona verde.
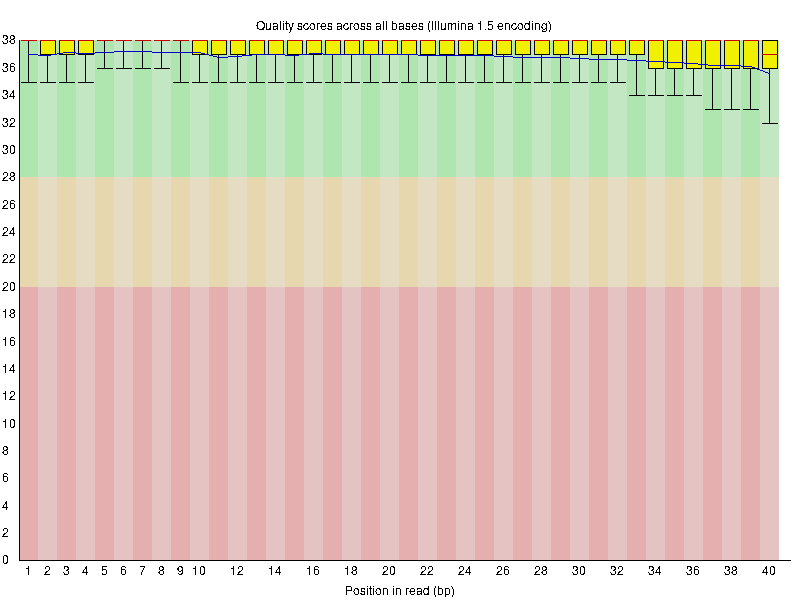
- Visualiza el contenido del archivo “_files/secuencias_bash.fastq” empleando head.
head _files/secuencias_bash.fastq - Calcula el numero de lineas contenidas en este archivo.
wc -l _files/secuencias_bash.fastq - Imprime las lineas 4 a 8 del archivo.
sed -n '4,8p' _files/secuencias_bash.fastq - Podemos utilizar la función
grep -cpara contar el número de secuencias de un archivo (de nuevo, sustituya archivo por el nombre de uno de los archivos):grep -c "^@SRR" _files/secuencias_bash.fastq