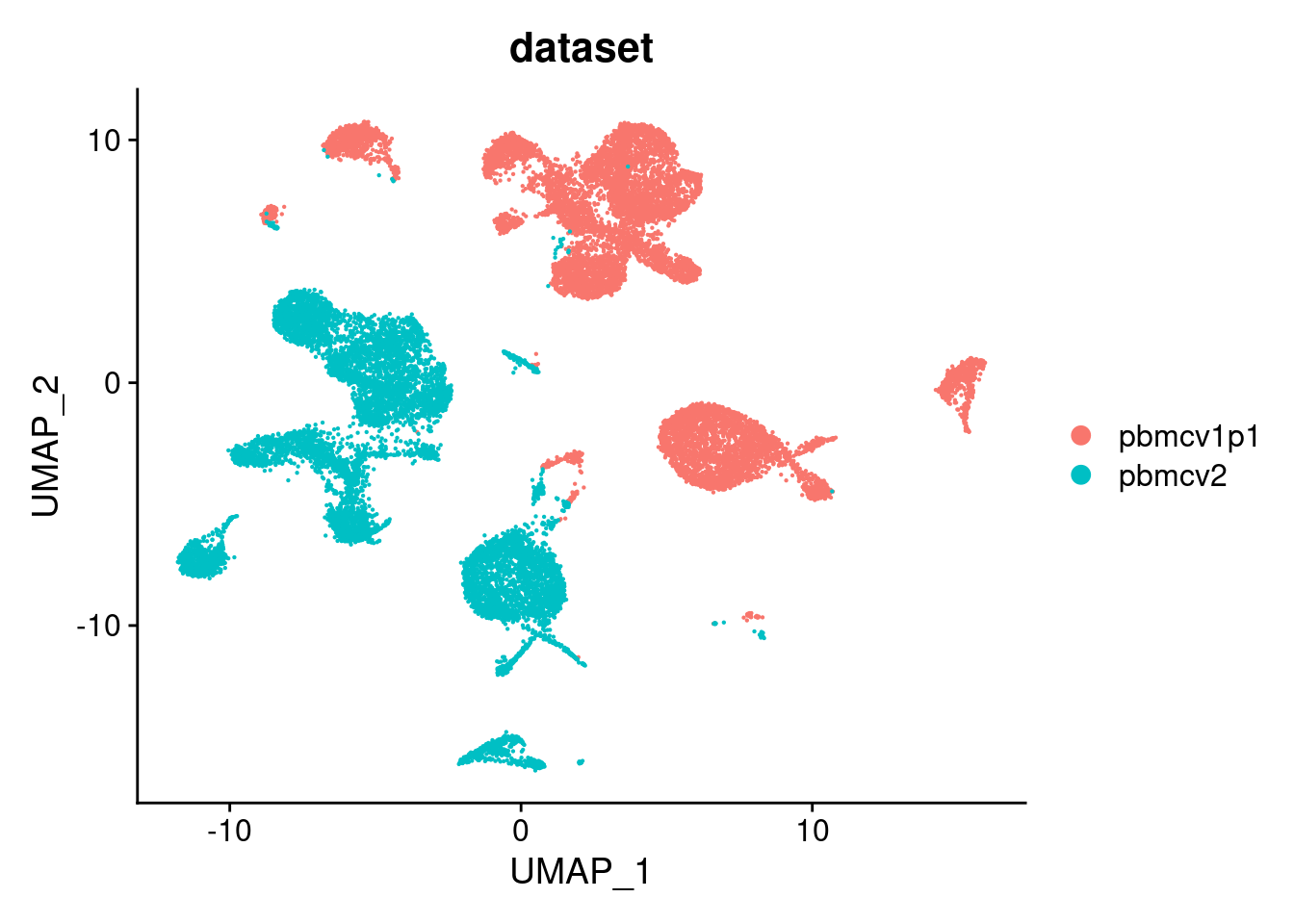
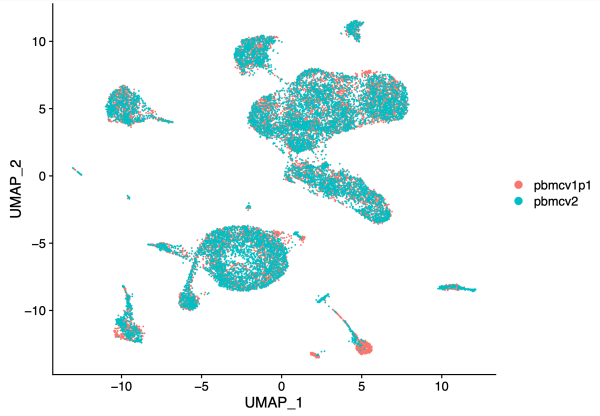
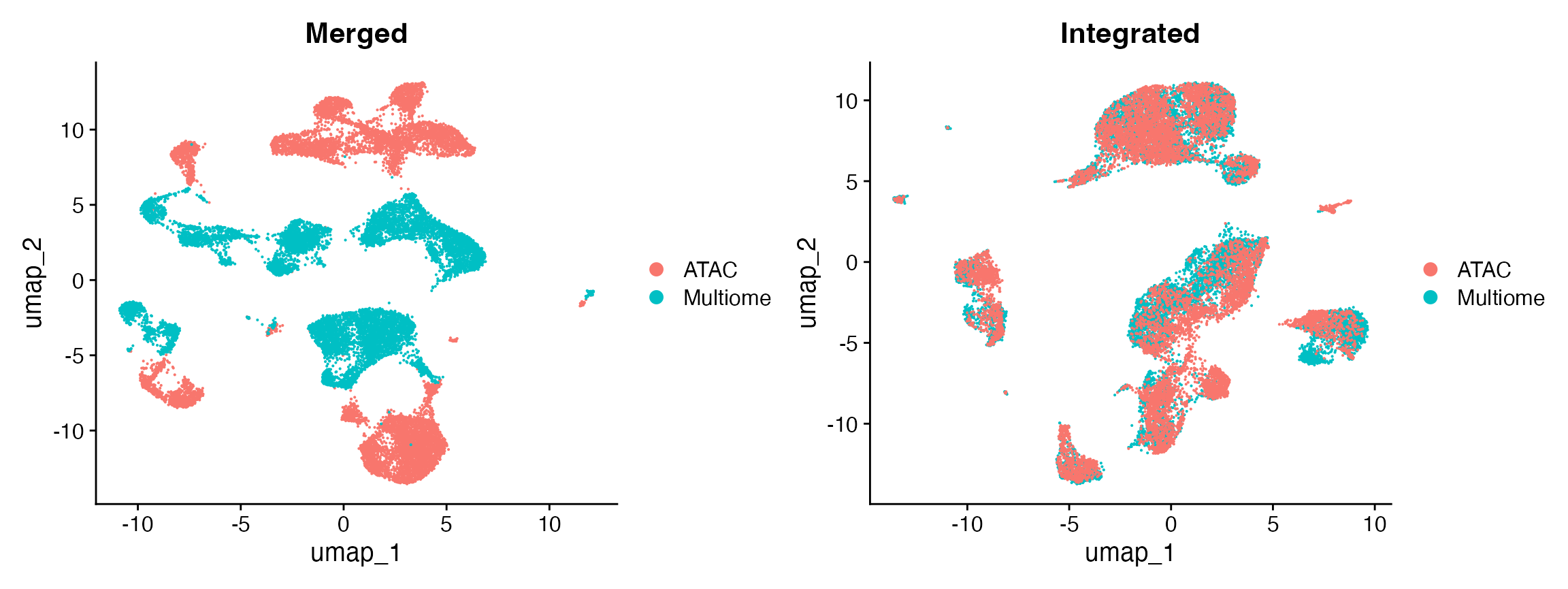
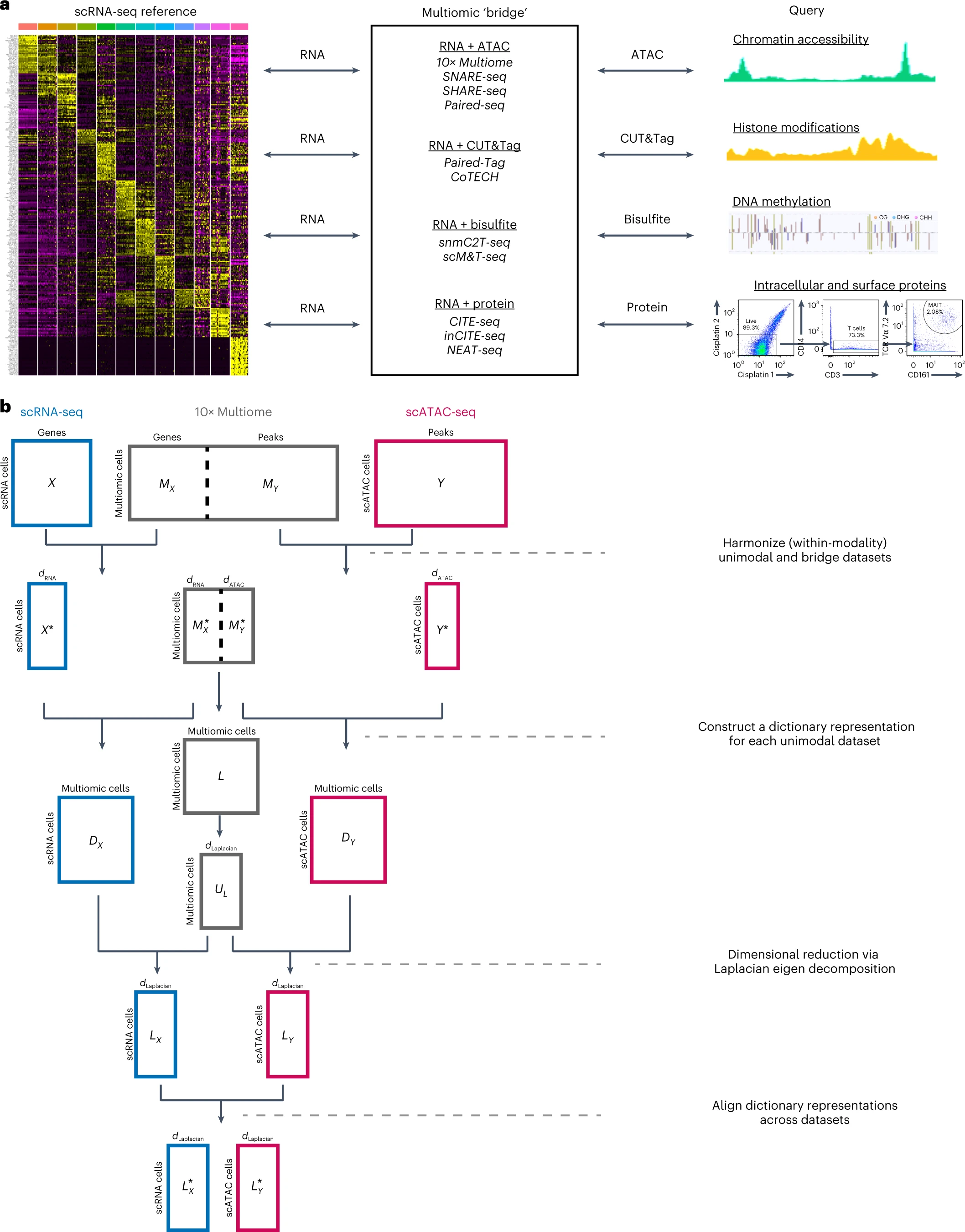
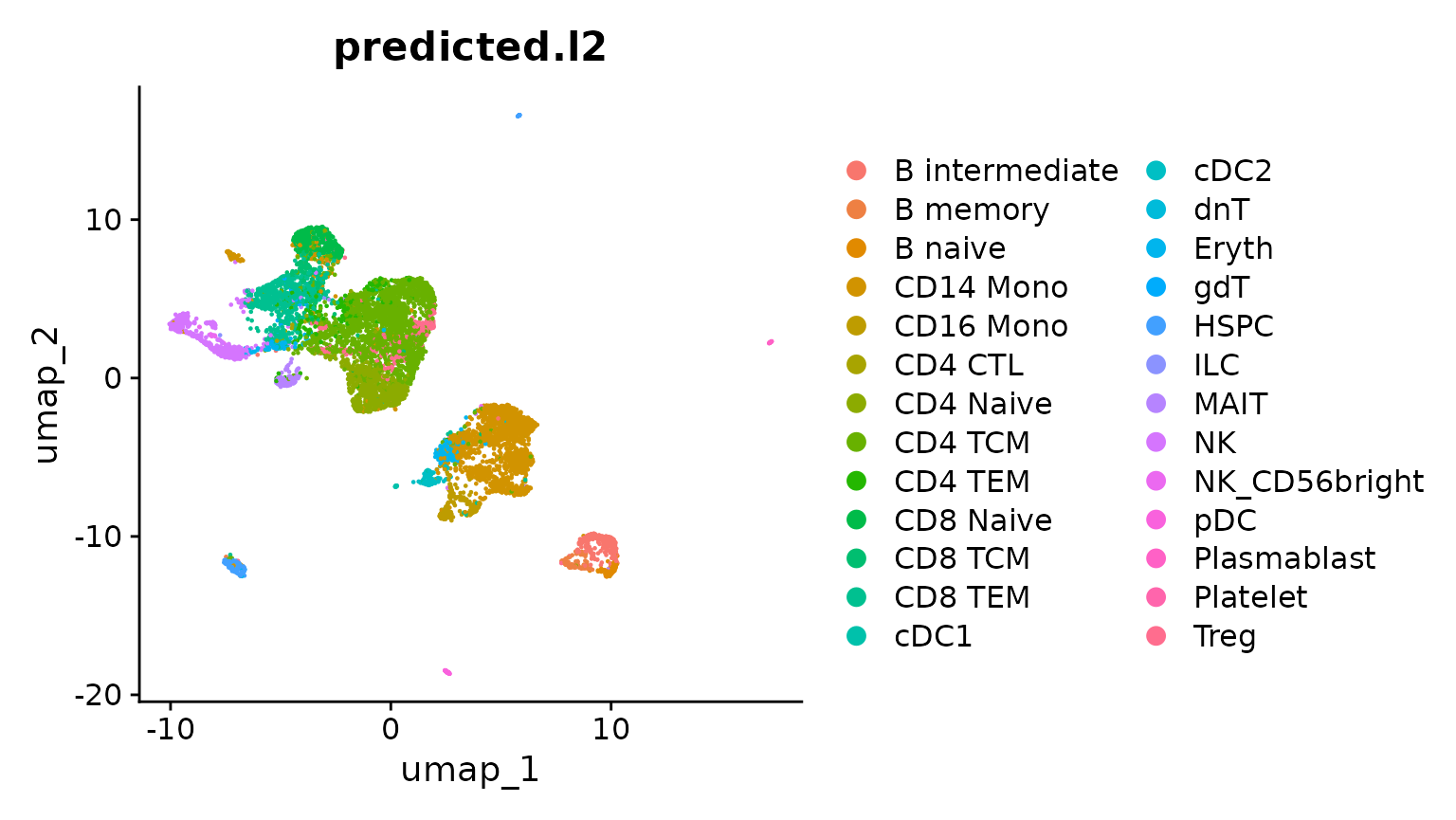
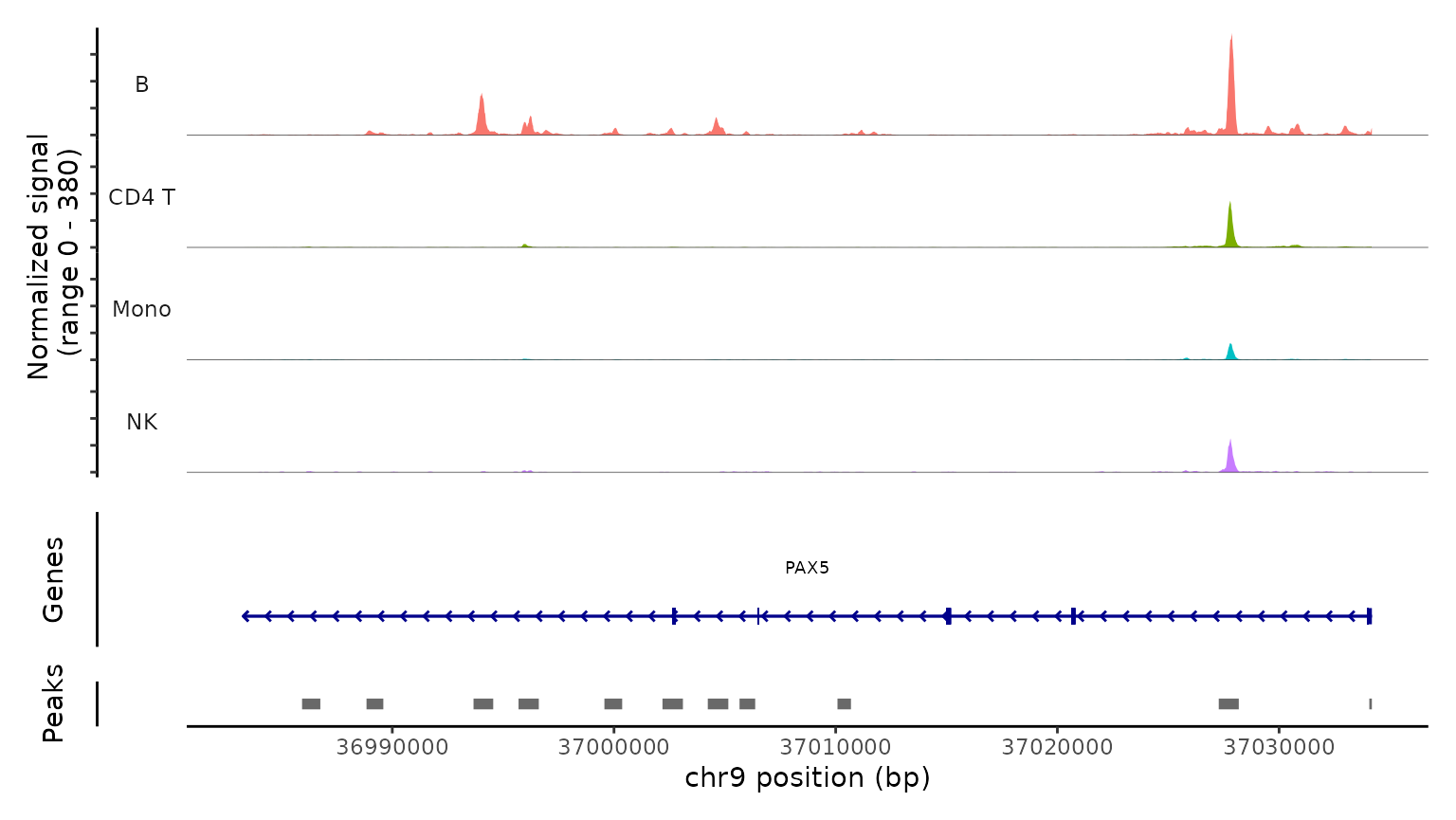
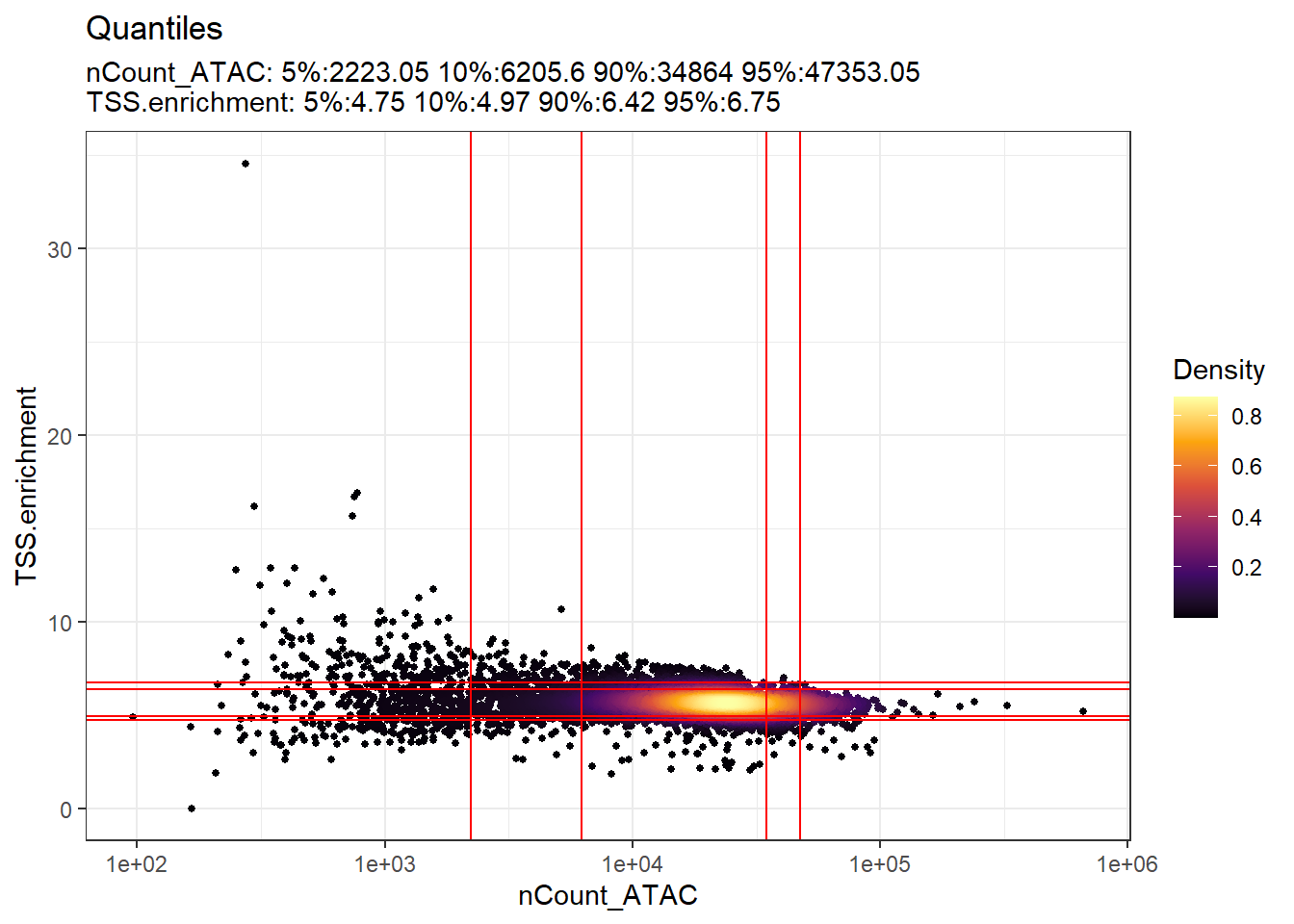
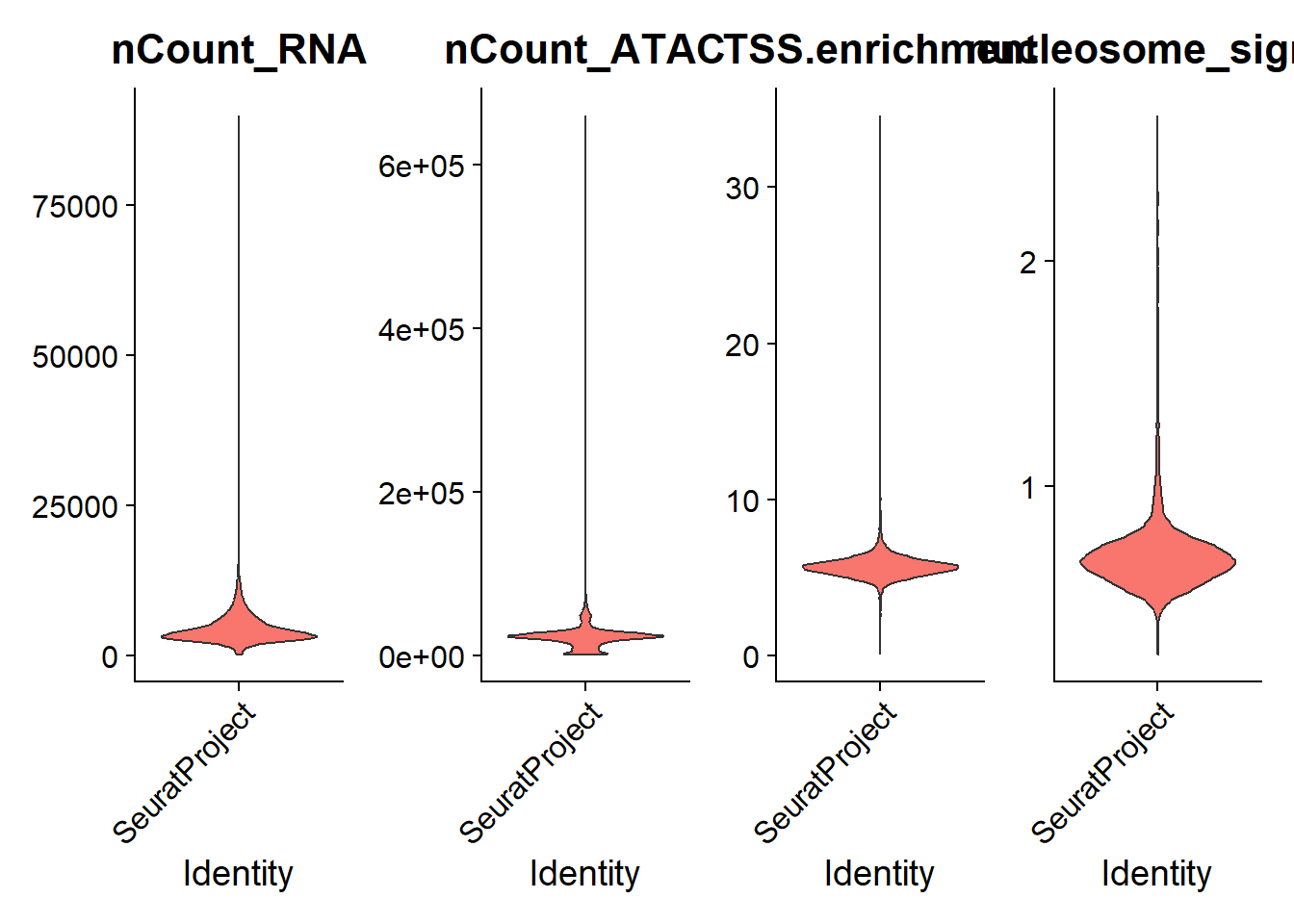
PC_ 1
Positive: VCAN, SAT1, PLXDC2, SLC8A1, NEAT1, DPYD, ZEB2, NAMPT, LRMDA, LYZ
FCN1, AOAH, LYN, ANXA1, CD36, JAK2, ARHGAP26, TYMP, RBM47, PID1
PLCB1, LRRK2, ACSL1, LYST, IRAK3, DMXL2, MCTP1, TCF7L2, AC020916.1, GAB2
Negative: EEF1A1, RPL13, RPS27, RPL13A, RPL41, RPS27A, RPS2, RPL3, RPS12, RPL11
RPS18, BCL2, LEF1, BCL11B, RPL23A, RPL10, BACH2, IL32, RPL34, INPP4B
RPS3, CAMK4, IL7R, RPS26, RPS14, RPL30, RPL19, LTB, RPLP2, SKAP1
PC_ 2
Positive: GNLY, CCL5, NKG7, CD247, IL32, PRKCH, PRF1, BCL11B, INPP4B, LEF1
VCAN, IL7R, DPYD, THEMIS, GZMA, NEAT1, CAMK4, RORA, AOAH, PLCB1
TXK, GZMH, TC2N, TRBC1, ANXA1, FYB1, STAT4, PITPNC1, TRAC, AAK1
Negative: BANK1, IGHM, AFF3, IGKC, RALGPS2, MS4A1, CD74, PAX5, EBF1, FCRL1
CD79A, OSBPL10, LINC00926, COL19A1, BLK, NIBAN3, IGHD, CD22, HLA-DRA, CD79B
ADAM28, PLEKHG1, AP002075.1, COBLL1, LIX1-AS1, CCSER1, TCF4, BCL11A, STEAP1B, GNG7
PC_ 3
Positive: LEF1, CAMK4, EEF1A1, MAML2, PDE3B, FHIT, BACH2, IL7R, INPP4B, TSHZ2
SERINC5, NELL2, CCR7, BCL2, ANK3, PRKCA, FOXP1, TCF7, BCL11B, AC139720.1
RPL11, LTB, RPL13, OXNAD1, RPS27A, MLLT3, TRABD2A, RPS12, RASGRF2, VCAN
Negative: GNLY, NKG7, CCL5, PRF1, GZMA, GZMH, KLRD1, SPON2, FGFBP2, CST7
GZMB, CCL4, TGFBR3, KLRF1, CTSW, BNC2, PDGFD, ADGRG1, PPP2R2B, PYHIN1
A2M, IL2RB, CLIC3, MCTP2, NCAM1, ACTB, TRDC, SAMD3, NCALD, MYBL1
PC_ 4
Positive: BANK1, IGHM, VCAN, IGKC, PAX5, MS4A1, GNLY, FCRL1, RALGPS2, EBF1
LINC00926, CD79A, OSBPL10, COL19A1, NKG7, ARHGAP24, CCL5, IGHD, BACH2, PLCB1
PDE4D, DPYD, LIX1-AS1, CD22, LARGE1, PLEKHG1, PLXDC2, STEAP1B, ANXA1, AP002075.1
Negative: TCF4, LINC01374, CUX2, AC023590.1, RHEX, PLD4, EPHB1, LINC01478, PTPRS, LINC00996
ZFAT, LILRA4, CLEC4C, COL26A1, FAM160A1, CCDC50, PLXNA4, ITM2C, SCN9A, COL24A1
UGCG, RUNX2, NRP1, GZMB, IRF8, SLC35F3, JCHAIN, BCL11A, TNFRSF21, AC007381.1
PC_ 5
Positive: CDKN1C, FCGR3A, TCF7L2, IFITM3, MTSS1, AIF1, CST3, LST1, MS4A7, WARS
PSAP, ACTB, SERPINA1, HLA-DPA1, IFI30, CD74, COTL1, FCER1G, CFD, HLA-DRA
FTL, HLA-DRB1, SMIM25, CSF1R, TMSB4X, FMNL2, SAT1, MAFB, S100A4, HLA-DPB1
Negative: VCAN, TCF4, PLCB1, LINC01374, RHEX, CUX2, AC023590.1, DPYD, EPHB1, LINC01478
PTPRS, ANXA1, ZFAT, LINC00996, CD36, GZMB, PLXDC2, ARHGAP26, FCHSD2, COL26A1
CLEC4C, LILRA4, ACSL1, PLXNA4, AC020916.1, DYSF, ARHGAP24, CSF3R, LRMDA, MEGF9