The previous analysis (Practical 14 - Find differentially accessible peaks between cell types, da_peaks) can help us to identify putative cis-regulatory elements that are critical for regulating cell type identity or cell state transitions. This is usually achieved via the binding of certain trans-regulators, e.g. TFs, to those open chromatin regions.
The binding of most TFs have strong sequence specificity, which can be summarized into sequence motifs, i.e. the TF binding motifs. If certain TFs play important roles in those regulations, it is very likely that the cell-type-specific peaks are regions enriched for the binding, with their genomic sequences enriched for the corresponding TF binding motifs. In this case, by checking the motifs enriched in those cell-type-specific peaks, we may be then able to identify TFs responsible for establishment and maintanence of cell type identity.
To do that, we also need a database of TF binding motifs. Indeed, there are several commonly used databases, including the commercial ones like TRANSFAC and open source ones like JASPAR. By scanning for sequences matching those motifs, we can predict possible binding sites of the TFs with binding motif information in the databases, and then check their enrichment in the peak list of interest in relative to the other peaks.
9.0.1 The Motif Class
The Motif class stores information needed for DNA sequence motif analysis, and has the following slots:
data: a sparse feature by motif matrix, where entries are 1 if the feature contains the motif, and 0 otherwise
pwm: A named list of position weight or position frequency matrices
motif.names: a list of motif IDs and their common names
positions: A GRangesList object containing the exact positions of each motif
meta.data: Additional information about the motifs
Many of these slots are optional and do not need to be filled, but are only required when running certain functions. For example, the positions slot will be needed if running TF footprinting. For more details on the Motif class.
9.0.2 Step 1: Adding motif information to the Seurat object
To add the DNA sequence motif information required for motif analyses, we can run the AddMotifs() function:
Code
library(Signac)library(Seurat)# BiocManager::install("JASPAR2020")library(JASPAR2020) # Data package for JASPAR database (version 2020)# BiocManager::install("TFBSTools")library(TFBSTools) # Software Package for Transcription Factor Binding Site (TFBS) Analysislibrary(BSgenome.Hsapiens.UCSC.hg38)library(patchwork)# BiocManager::install("motifmatchr")library(motifmatchr)library(GenomicRanges)# BiocManager::install("ggseqlogo")library(ggseqlogo)
Code
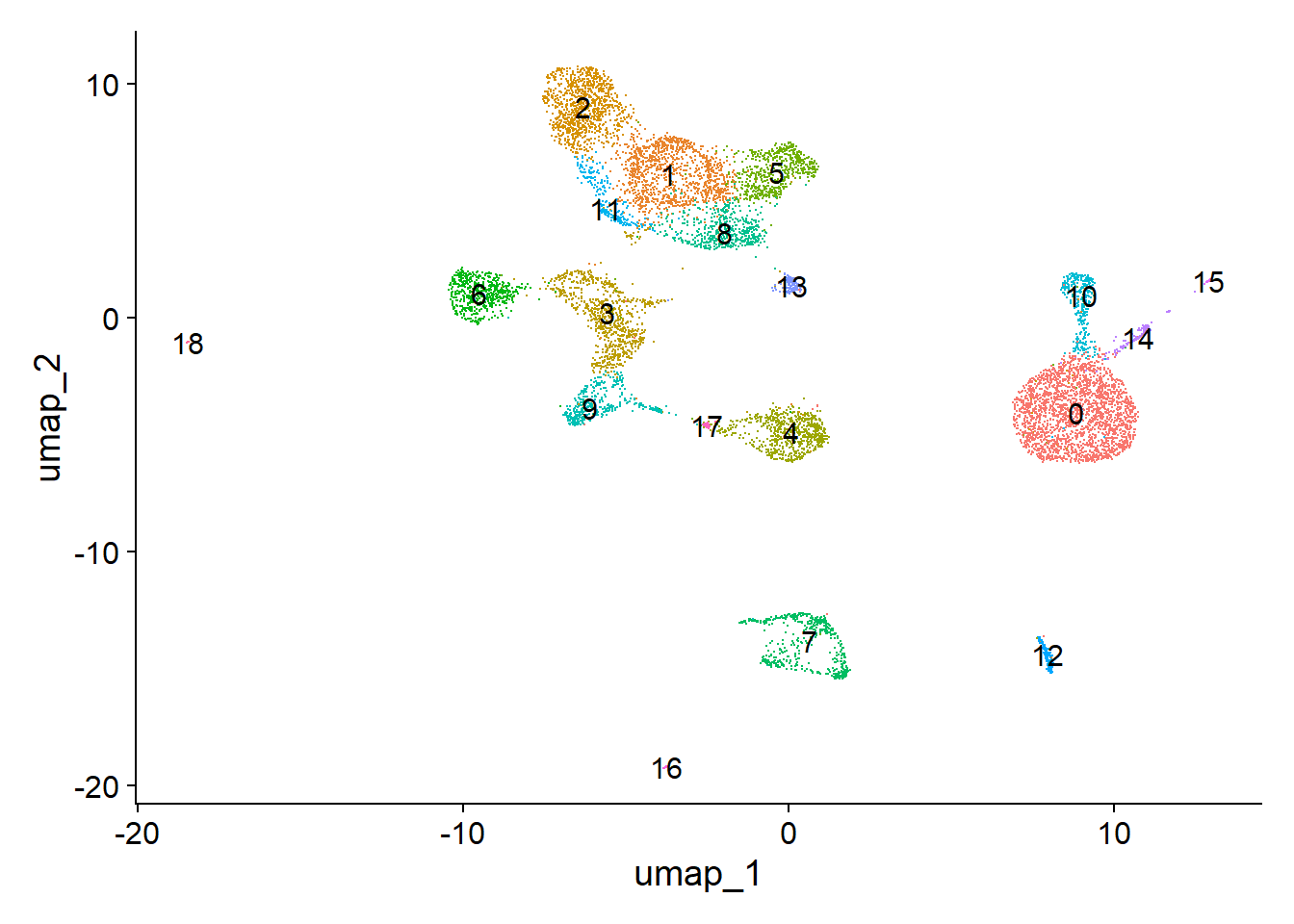
# load from Section 2 P14 - STEP: Find differentially accessible peaks between cell types, da_peaksload(file ="data/pbmc.RData") # from Practical 13# load(file = "data/pbmc_edited.RData")# Idents(pbmc) <- pbmc$predicted.idp1 <-DimPlot(pbmc, label =TRUE, pt.size =0.1) +NoLegend()p1
Code
# Get a list of motif position frequency matrices from the JASPAR databasepfm <-getMatrixSet(x = JASPAR2020,opts =list(species ="Homo sapiens", all_versions =FALSE))# add motif informationpbmc <-AddMotifs(object = pbmc,genome = BSgenome.Hsapiens.UCSC.hg38,pfm = pfm)
Building motif matrix
Finding motif positions
Creating Motif object
To facilitate motif analysis in Signac, we have created the Motif class to store all the required information, including a list of position weight matrices (PWMs) or position frequency matrices (PFMs) and a motif occurrence matrix. Here, the AddMotifs() function constructs a Motif object and adds it to our mouse brain dataset, along with other information such as the base composition of each peak. A motif object can be added to any Seurat assay using the SetAssayData() function. See the object interaction vignette for more information.
Cluster composition shows many clusters unique to the whole blood dataset:
To identify potentially important cell-type-specific regulatory sequences, we can search for DNA motifs that are overrepresented in a set of peaks that are differentially accessible between cell types.
Here, we find differentially accessible peaks between Pvalb and Sst inhibitory interneurons. For sparse data (such as scATAC-seq), we find it is often necessary to lower the min.pct threshold in FindMarkers() from the default (0.1, which was designed for scRNA-seq data).
We then perform a hypergeometric test to test the probability of observing the motif at the given frequency by chance, comparing with a background set of peaks matched for GC content.
Code
# Find differentially accessible peaks in cluster 0 compared to cluster 1 (~1h 7m)da_peaks <-FindMarkers(object = pbmc,#ident.1 = 'CD14+ Monocytes',#ident.2 = 'pre-B cell',ident.1 ='0',ident.2 ='1',only.pos =TRUE,min.pct =0.05,test.use ='LR',latent.vars ='nCount_peaks')
Code
# Get the top differentially accessible peaks, with lowest p-valuestop.da.peak <-rownames(da_peaks[da_peaks$p_val <0.005& da_peaks$pct.1>0.2, ])
Code
# savesave(da_peaks, file ="data/pbmc_markers.RData")# save informationsave(pbmc, top.da.peak, file ="data/pbmc_motifs.RData")
Code
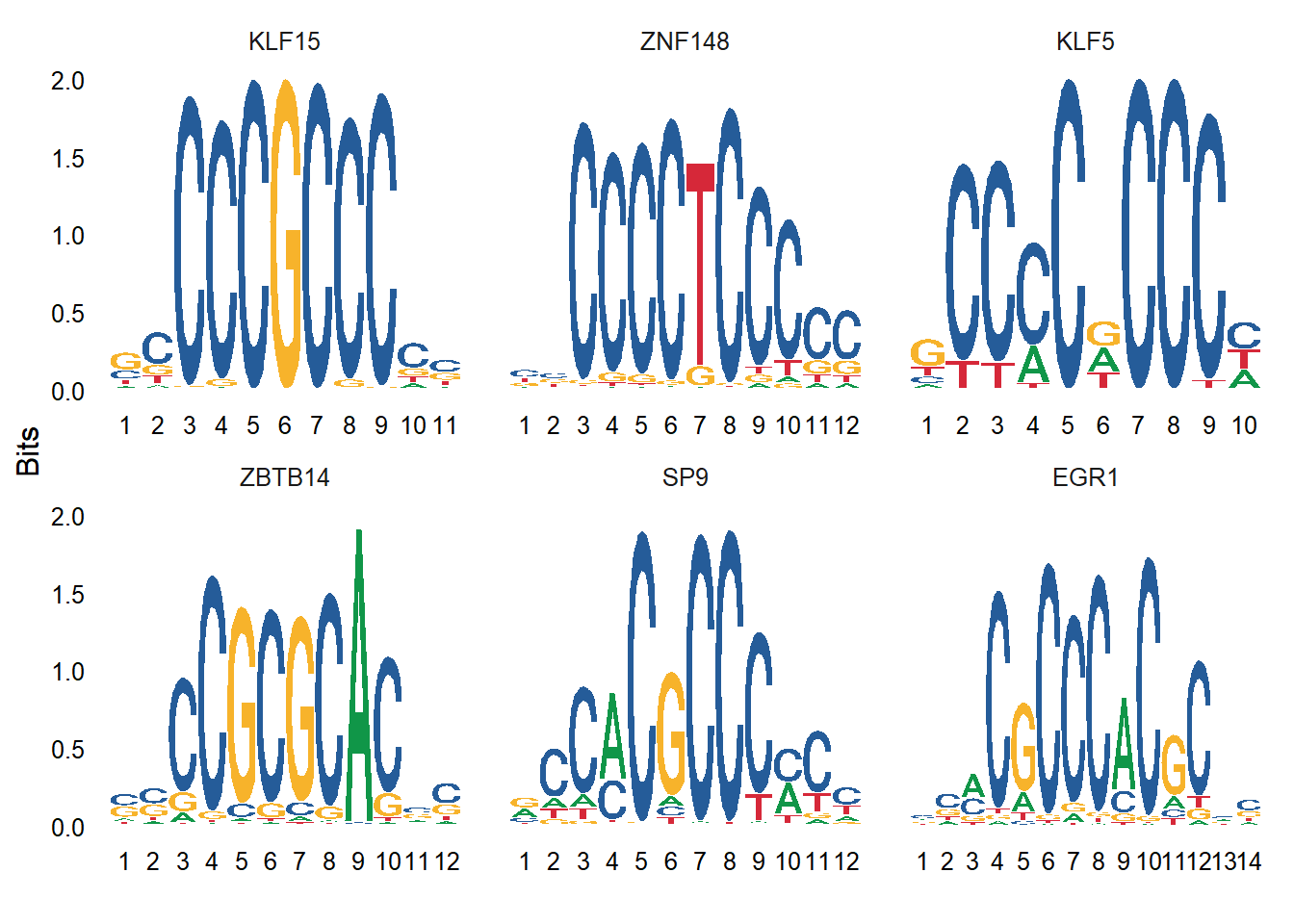
load(file ="data/pbmc_motifs.RData")# Find motifs enriched in these top differentially accessible peaksenriched.motifs <-FindMotifs(object = pbmc,features = top.da.peak)
Selecting background regions to match input sequence characteristics
Warning: The `<scale>` argument of `guides()` cannot be `FALSE`. Use "none" instead as
of ggplot2 3.3.4.
ℹ The deprecated feature was likely used in the ggseqlogo package.
Please report the issue at <https://github.com/omarwagih/ggseqlogo/issues>.
9.0.4Step 3: Motif activity scores
We can also compute a per-cell motif activity score by running chromVAR. The motif activity score for a motif M is based on the number of reads mapping to peaks with motif M, after normalization correction for various biases: GC content, average number of reads mapping across all cells etc. You can read more about chromVar here. Motif activity scores allow us to visualize motif activities per cell.
Note
ChromVAR identifies motifs associated with variability in chromatin accessibility between cells. See the chromVAR paper for a complete description of the method.
It is also possible to directly test for differential activity scores between cell types, without looking at peaks with differential binding. This tends to give similar results as performing an enrichment test on differentially accessible peaks between the cell types (shown above).
Caution
This takes a while to run (around 5 minutes on Uppmax).
# Use chromVAR to calculate the motif activities of all motifs in all cells.chromvar_pbmc <-RunChromVAR(object = pbmc,genome = BSgenome.Hsapiens.UCSC.hg38,verbose =TRUE)DefaultAssay(chromvar_pbmc) <-'chromvar'# Look at resultspbmc$chromvarGetAssayData(pbmc$chromvar)[1:10,1:3]
Code
# Have a look at the activitiy of the FOS motif, which has id MA0476.1DefaultAssay(pbmc) <-'chromvar'FeaturePlot(object = pbmc,features ="MA0476.1",min.cutoff ='q10',max.cutoff ='q90',pt.size =0.1)p1 + p2
We can also directly test for differential activity scores between cell types. This tends to give similar results as performing an enrichment test on differentially accessible peaks between the cell types (shown above).
When performing differential testing on the chromVAR z-score, we can set mean.fxn=rowMeans and fc.name="avg_diff" in the FindMarkers() function so that the fold-change calculation computes the average difference in z-score between the groups.
Code
# Look for motifs that have differential activity between clusters 0 and 1.differential.activity <-FindMarkers(object = pbmc,ident.1 ='0',ident.2 ='1',only.pos =TRUE,test.use ='LR',min.pct =0.2,latent.vars ='nCount_peaks')MotifPlot(object = pbmc,motifs =head(rownames(differential.activity)),assay ='peaks')
10 Building trajectories with Monocle 3
Also you can construct cell trajectories with Monocle 3 using single-cell ATAC-seq data. Please see the Monocle 3 website for information about installing Monocle 3.